Hereditäre ATTR-Amyloidose: eine lebensbedrohliche, Multisystemerkrankung1-4
Die Erkrankung betrifft mehrere Organe und führt zu unterschiedlichen Symptomen1,3,4
Da Amyloidfibrillen im Gewebe im gesamten Körper, einschließlich Nerven, Herz und Magen-Darm-Trakt, abgelagert werden, erstrecken sich die Symptome der an hATTR-Amyloidose erkrankten Patienten über ein breites Spektrum, hierunter sensorische und motorische, autonome und kardiale Symptome.1-5
Tatsächlich leidet mehr als die Hälfte der Patienten mit hATTR an einem gemischten Phänotyp.6,7
Konstellation möglicher Anzeichen und Symptome von hereditärer ATTR-Amyloidose
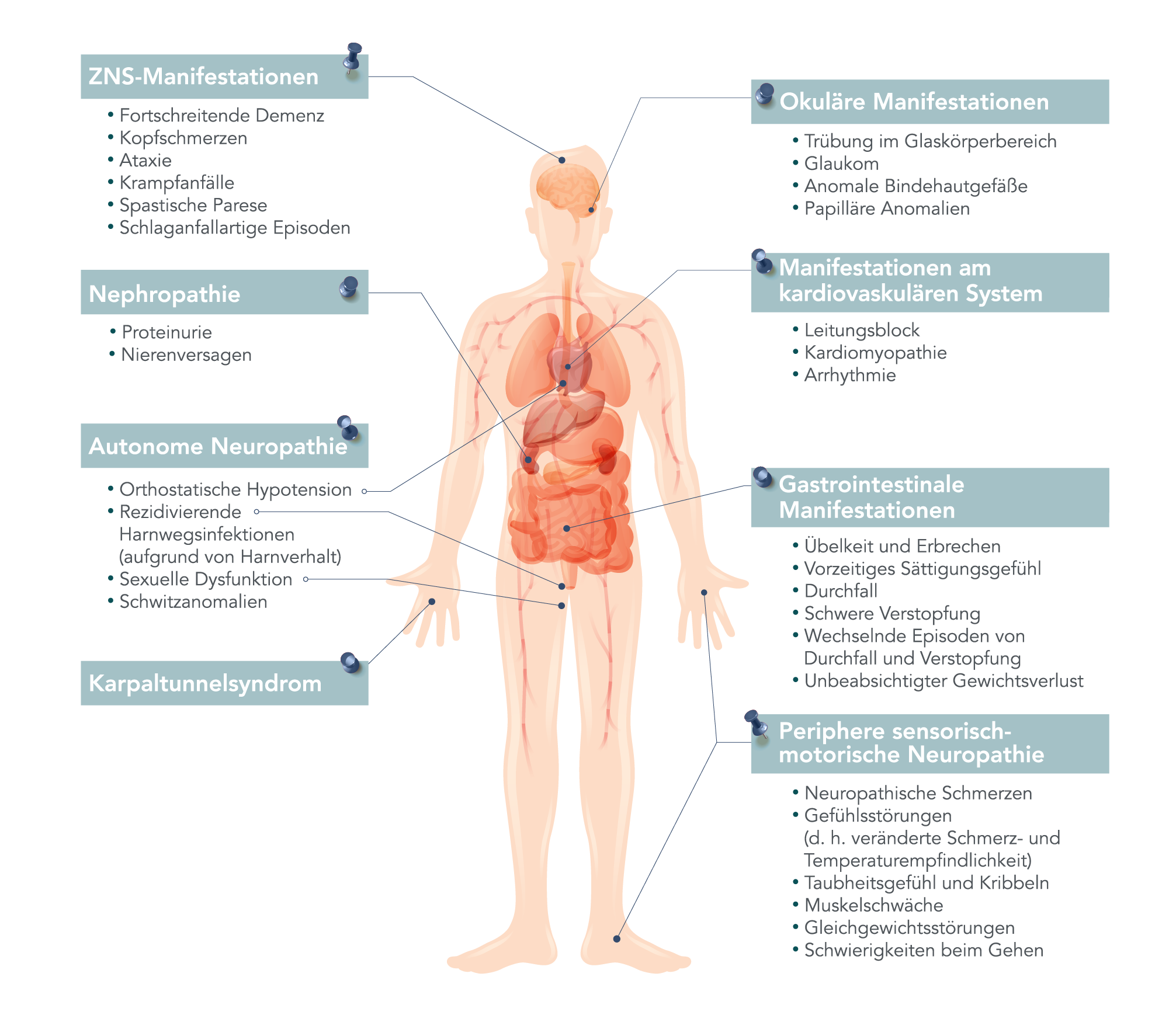
Adaptiert von Conceição I, et al. J Peripher Nerv Syst. 2016;21(1):5-9.
Die Symptome sind selbst bei Patienten mit derselben Mutation sehr uneinheitlich. Bei Patienten in derselben Familie können die Symptome stark variieren8
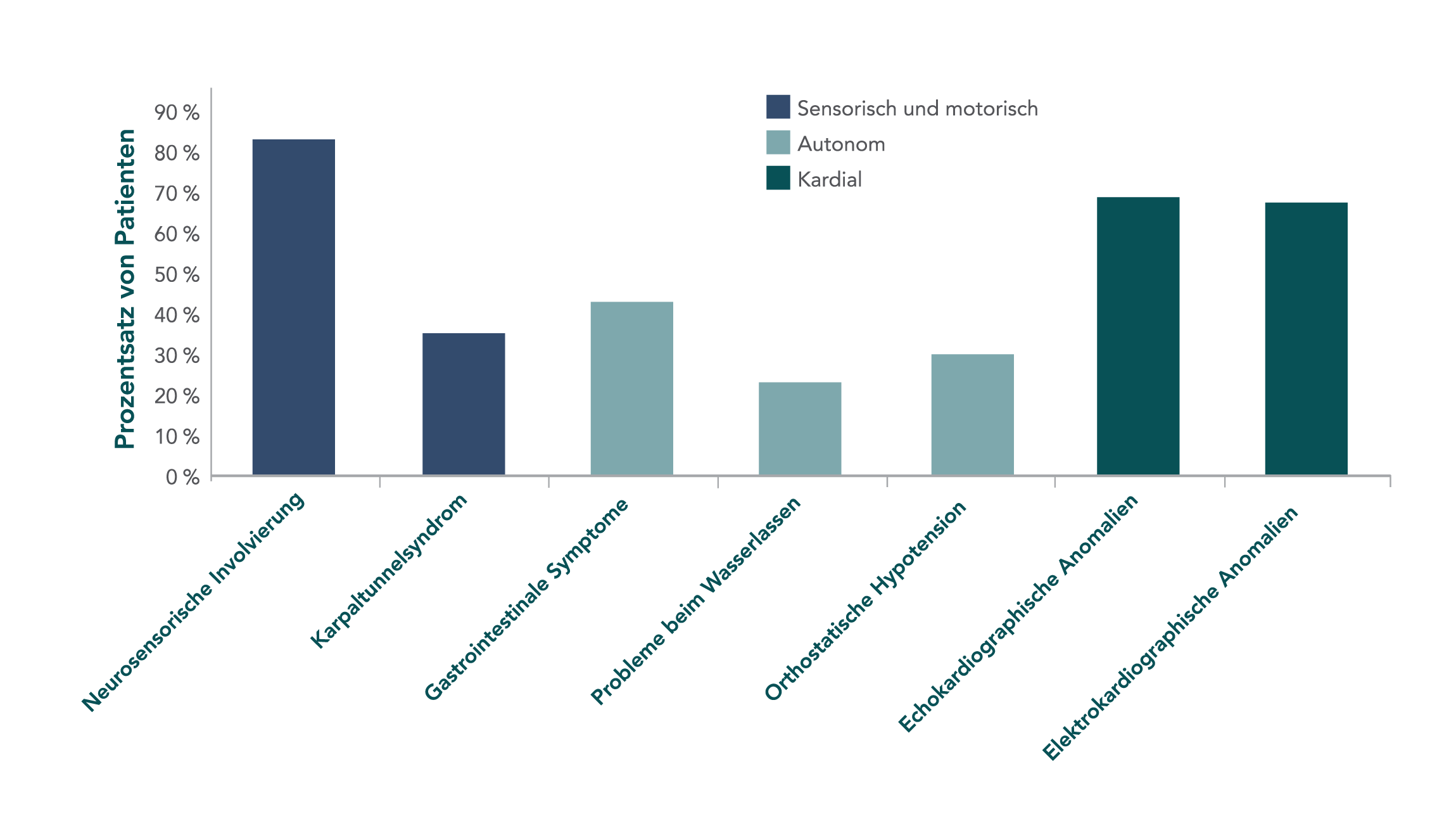
Klinische Baseline-Merkmale von 186 Patienten mit vererbbarer ATTR-Amyloidose in einer multizentrischen Studie.6